Mutations of potential epidemiological relevance
Published: 09 February 2021
IMPORTANT: while these are up to date, our analyses cannot offer a complete overview of the current situation in our country. Available data are fragmented and do not have the same level of completeness or accuracy for different geographic regions. The information reported in this analysis is purely descriptive and does not necessarily represent the real situation in Italy.
In other words, it is like looking at a very low-res picture: details are blurred and it is nearly impossible to understand the real situation with a sufficient degree of precision. Furthermore, it is like some bits of the picture have been shot in spring, a few in summer and fall and some more in winter. That is, we have both spatial and temporal blurriness. Improving the resolution of this picture both at the spatial and temporal level, that is increasing the number of viral genomes regularly sequenced and shared in public repositories in our country, is one of the most ambitious objectives of this portal.
Glossary
The genome of SARS-CoV-2, the virus that causes COVID-19 is made up of an RNA molecule composed by approximately 30,000 ribonucleotides. Each of these nucleotides and their disposition along the molecule can be described using a four letters code, with each letter corresponding to one of the four possible nucleobases: A (Adenine), C (Cytosine), U (Uracil) or G (Guanine). The exact and complete succession of these letters determines the “genome sequence” of the virus. The term “lineage” is used to indicate a type of the viral genome that differs in one or more positions from the “reference genome” (the first available genome sequence of SARS-CoV-2). Differences between genomic sequences can be substitutions (the most common, we find one letter instead of another), insertions (a bit of sequence more than the reference), or deletions (a bit of sequence less than the reference). Each of the nucleotides that make up the genome of the virus can change over time, both due to evolution and selection, but also (in most cases) in a completely random manner. Each change of a single ribonucleotide of the RNA genome of the virus is called a “mutation”.
After taking a vaccine, or following a previous infection, our immune system develops antibodies that can recognize specific portions of the viral proteins and trigger an immune response. This mechanism protects us from a second infection.
Specific mutations that change the properties of some viral proteins, can make our antibodies less effective in recognizing the virus. These types of mutations could represent a potential source of risk, and result in a decrease in the effectiveness of currently available vaccines.
Mutations associated with lineages of epidemiological interest
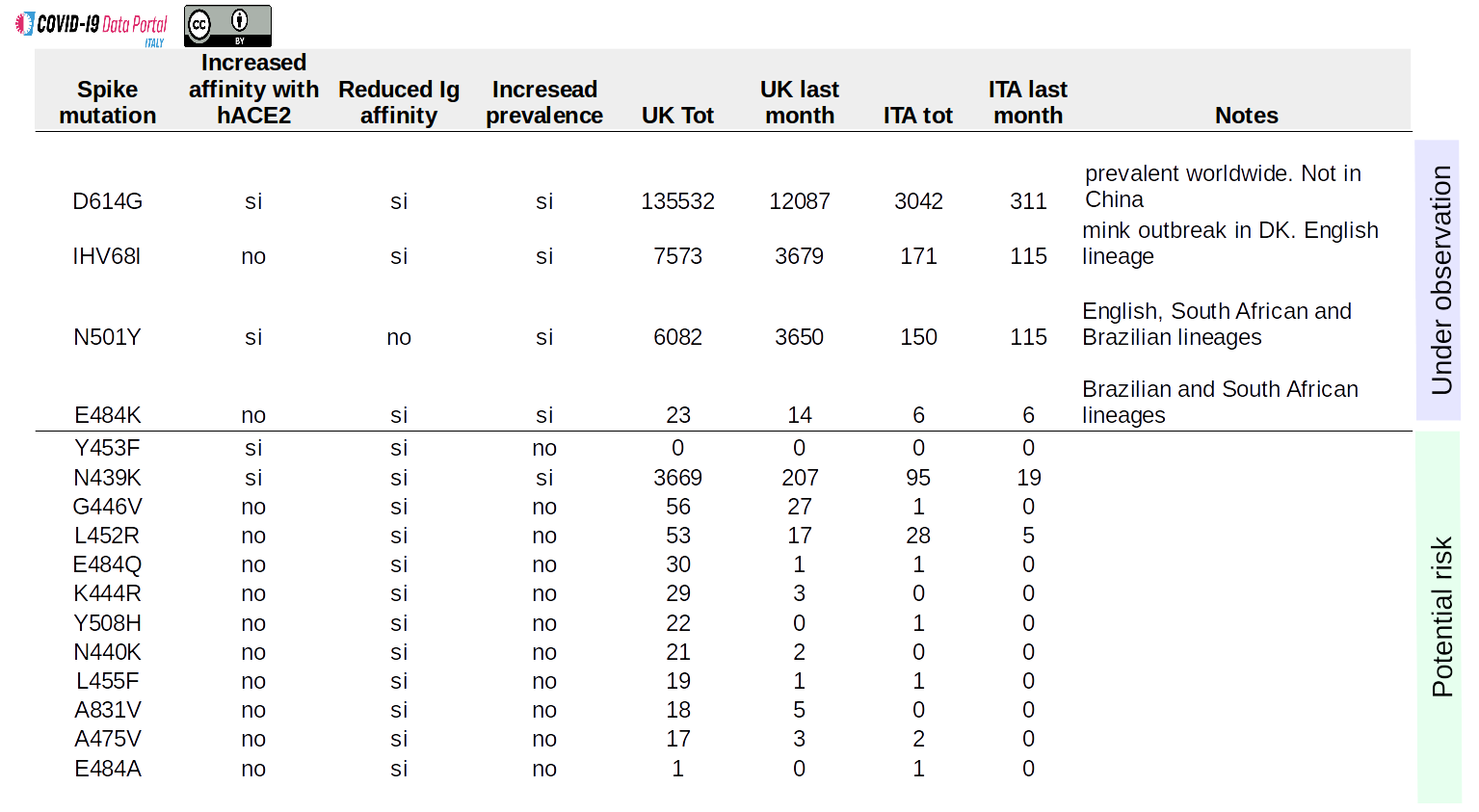
This report contains a list of the mutations associated with viral lineages of potential epidemiological interest. For a more detailed description of these lineages, see the highlight SARS-CoV-2 lineages from Feb 5 2021.
All of the mutations herein reported are associated with changes in the Spike glycoprotein, the viral protein that can recognize our cells and allows SARS-CoV-2 to infect them. Due to its importance, Spike is one of the main targets of our immune system, and several human antibodies are able to recognize it, thus neutralizing the ability of the virus to enter our cells. For these reasons, Spike glycoprotein is the natural target for the development of vaccines. Specific mutations associated with changes in this protein can decrease the ability of our antibodies to recognize SARS-CoV-2, allowing the virus to infect even people who have already been vaccinated or infected.
Other mutations in Spike can make the protein more efficient in recognizing its human host, and create “more infectious” forms of the virus. For this reason, the study of mutations in the Spike glycoprotein is crucial to identify novel, potentially dangerous, lineages of the virus.
Mutations associated with lineages of epidemiological relevance
The D614G mutation was not present in the genome of the virus when SARS-CoV-2 was first isolated, but it is now almost ubiquitous. It is found in numerous variants and appears to be associated with a moderate increase in the transmissibility of SARS-CoV-2 [4].
The N501Y mutation initially identified in the United Kingdom [5], is associated with several lineages of the virus that are considered to have increased transmissibility, such as the English (B.1.1.7), the Brazilian (P.1) and South African (501Y.V2) variants. [6]. It is believed that this mutation may favor the binding of the virus to the human receptor protein.
E484K is specific to the Brazilian (P.1) and South African (501Y.V2) variants. Preliminary studies suggest that this mutation can shield the virus from being recognized by some antibodies [7]. Early analyses carried out in Brazil suggest a higher prevalence of this mutation in cases of re-infection. These data are however to be considered preliminary [8].
IHV68I is a deletion, a mutation associated with the loss of 2 amino acids in the Spike glycoprotein. IHV68I has appeared several times “independently” in the genome of different variants of SARS-CoV-2. It was first identified in immuno-suppressed patients treated with monoclonal antibodies [9], but it is also associated with a zoonotic event of transmission from human to minks and then from minks back to human, which caused a recent epidemic in Denmark [10]. This deletion is also found in the English variant B.1.1.7. It is believed to decrease the ability of our antibodies to recognize the virus [11]
Other mutations
Mutations marked by the “potential risk” flag are not associated with any particular lineage of epidemiological interest but have particular features that make them noteworthy. These are reported only for the sake of completeness and to facilitate future comparisons. At present they do not constitute a source of concern.
Among these, the N439K mutation is probably the most interesting one. Preliminary studies indicate that this particular mutation is able to both improve the ability of SARS-CoV-2 to recognize the hACE2 receptor protein, while at the same time making the virus less efficiently recognized by the human immune system [12]. Recently, several variants of SARS-CoV-2 have been found that carry both N439K and IHV68I in their genome [9]. The functional implications are currently unclear.
Source: https://www.gisaid.org/ (for genomic data)
Bibliography
[1] “Weekly epidemiological update -27January 2021” (https://www.who.int/publications/m/item/weekly-epidemiological-update—27-january-2021)
[2] “European Centre for Disease Prevention and Control. Risk related to spread of new SARS-CoV-2 variants of concern in the EU/EEA, first update –21 January 2021. ECDC: Stockholm; 2021.” (https://www.ecdc.europa.eu/sites/default/files/documents/COVID-19-risk-related-to-spread-of-new-SARS-CoV-2-variants-EU-EEA-first-update.pdf)
[3] “European Centre for Disease Prevention and Control. Sequencing of SARS-CoV-2: first update. 18 January 2021. ECDC: Stockholm; 2021. (https://www.ecdc.europa.eu/sites/default/files/documents/Sequencing-of-SARS-CoV-2-first-update.pdf)
[4] Korber, B., et al., Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell, 2020. 182(4): p. 812-827.e19
[5] Rambaut, A., et al. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. 2020; Available at: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineagein-the-uk-defined-by-a-novel-set-of-spike-mutations/563.
[6] Tegally, H., et al., Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv, 2020: p.2020.12.21.20248640.
[7] Weisblum, Y., et al., Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. bioRxiv, 2020: p. 2020.07.21.214759.
[8] Resende, P.C., et al. Spike E484K mutation in the first SARS-CoV-2 reinfection case confirmed in Brazil, 2020. 2021; Available at: https://virological.org/t/spike-e484k-mutation-in-the-first-sars-cov-2-reinfection-case-confirmed-in-brazil-2020/584
[9] Kemp, S., et al., Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/V70. bioRxiv, 2020: p. 2020.12.14.422555.
[10] Fonager J., et al., Working paper on SARS-CoV-2 spike mutations arising in Danish mink, their spread to humans and neutralization data. Available at https://files.ssi.dk/Mink-cluster-5-short-report_AFO2. 2020.
[11] Haynes, W.A., et al., Impact of B.1.1.7 variant mutations on antibody recognition of linear SARS-CoV2 epitopes. medRxiv, 2021: p. 2021.01.06.20248960.
[12] Thomson, E.C., et al., The circulating SARS-CoV-2 spike variant N439K maintains fitness while evading antibody-mediated immunity. Cell, 2021. Epub ahead of print https://doi.org/10.1016/j.cell.2021.01.037.