Mutazioni di potenziale interesse epidemiologico
Pubblicato: 09 February 2021
IMPORTANTE: queste analisi, per quanto aggiornate non possono offrire una panoramica completa della situazione nel nostro paese. Infatti, dati aggiornati sono disponibili in maniera frammentaria e solamente per alcune regioni. Le informazioni riportate in questa analisi hanno valore puramente descrittivo, e non necessariamente rappresentano la situazione reale.
In altre parole, è come guardare una fotografia a bassissima risoluzione: i dettagli sono sfocati ed è quasi impossibile sapere con un sufficiente grado di precisione quale sia la situazione reale. Per di più, è come se alcune parti della fotografia fossero state scattate in primavera, alcune in estate e in autunno e altre in inverno. Infatti, la maggior parte dei dati che abbiamo non sono allineati sia in termini geografici che temporali. Per migliorare la risoluzione dell’immagine, e ottenere un quadro più chiaro, in futuro sarà necessario sequenziare con regolarità un numero maggiore di sequenze genomiche e condividere tutti i dati in banche dati che permettano l’accesso senza alcun tipo di restrizione.
Glossario
Il genoma del virus SARS-CoV-2, l’agente eziologico del COVID-19 è composto da una molecola di RNA composta da circa 30.000 ribonucleotidi. Ciascuno di questi nucleotidi e il loro ordine lungo la molecola possono essere facilmente descritti utilizzando un codice composto di quattro lettere, che corrispondono alle quattro possibili basi azotate presenti in ciascun ribonucleotide: **A **(Adenina), C (Citosina), U (Uracile) oppure G (Guanina). La successione esatta e completa di queste lettere determina la “sequenza del genoma” del virus. Nel gergo comune, con il termine “variante” si tende ad indicare un tipo di genoma del virus che differisce in una o più posizioni rispetto a quella considerata come di “riferimento” (la sequenza del primo genoma di SARS-CoV-2 disponibile). Le differenze possono essere sostituzioni (le più comuni, troviamo una lettera al posto di un’altra), inserzioni (un pezzettino di sequenza in più rispetto al riferimento), o delezioni (un pezzettino di sequenza in meno rispetto al riferimento). Ciascuno dei nucleotidi che compongono il genoma del virus può cambiare nel tempo, sia a causa di processi di evoluzione e selezione, ma anche (nella maggior parte dei casi) in maniera completamente casuale. Ogni cambiamento di un singolo ribonucleotide del genoma a RNA del virus viene definito con il termine “mutazione”.
Dopo la vaccinazione, o in seguito ad una precedente infezione, il nostro sistema immunitario sviluppa degli anticorpi che sono in grado di riconoscere precise porzioni delle proteine del virus. Questo meccanismo è in genere in grado di proteggerci da una seconda infezione.
Particolari mutazioni possono cambiare le proprietà di alcune proteine virali, facendo in modo che queste siano riconosciute dai nostri anticorpi con minore efficienza. Questo tipo di mutazioni potrebbero rappresentare diminuire l’efficacia dei vaccini attualmente disponibili.
Mutazioni di interesse epidemiologico
Questo report riporta una lista di mutazioni associate a diverse varianti del virus, che sono ritenute di particolare interesse epidemiologico. Per una descrizione più puntuale di queste si rimanda al highlight Varianti SARS-CoV-2 del 05/02/2021.
Tutte le mutazioni qui riportate sono associate a cambiamenti nella glicoproteina Spike, la proteina virale che è in grado di riconoscere le nostre cellule e permette a SARS-CoV-2 di infettarle. Data l’importanza che riveste, Spike è una dei bersagli principali del nostro sistema immunitario, e diversi anticorpi umani sono in grado di riconoscerla, neutralizzando così la capacità del virus di entrare nelle nostre cellule. Proprio per questi motivi la glicoproteina Spike è il target naturale per lo sviluppo di vaccini.
Particolari mutazioni e/o cambiamenti nella proteina possono però diminuire la capacità dei nostri anticorpi di riconoscere il virus, conferendo al virus stesso la capacità di infettare anche persone già vaccinate o già infettate in precedenza.
Altre mutazioni di Spike possono rendere la proteina più efficiente nel riconoscere l’ospite umano, e creare forme del virus “più infettive”. Per questo motivo lo studio delle mutazioni di Spike è determinante per identificare nuove varianti del virus potenzialmente pericolose.
Descrizione delle varianti sotto osservazione in tabella
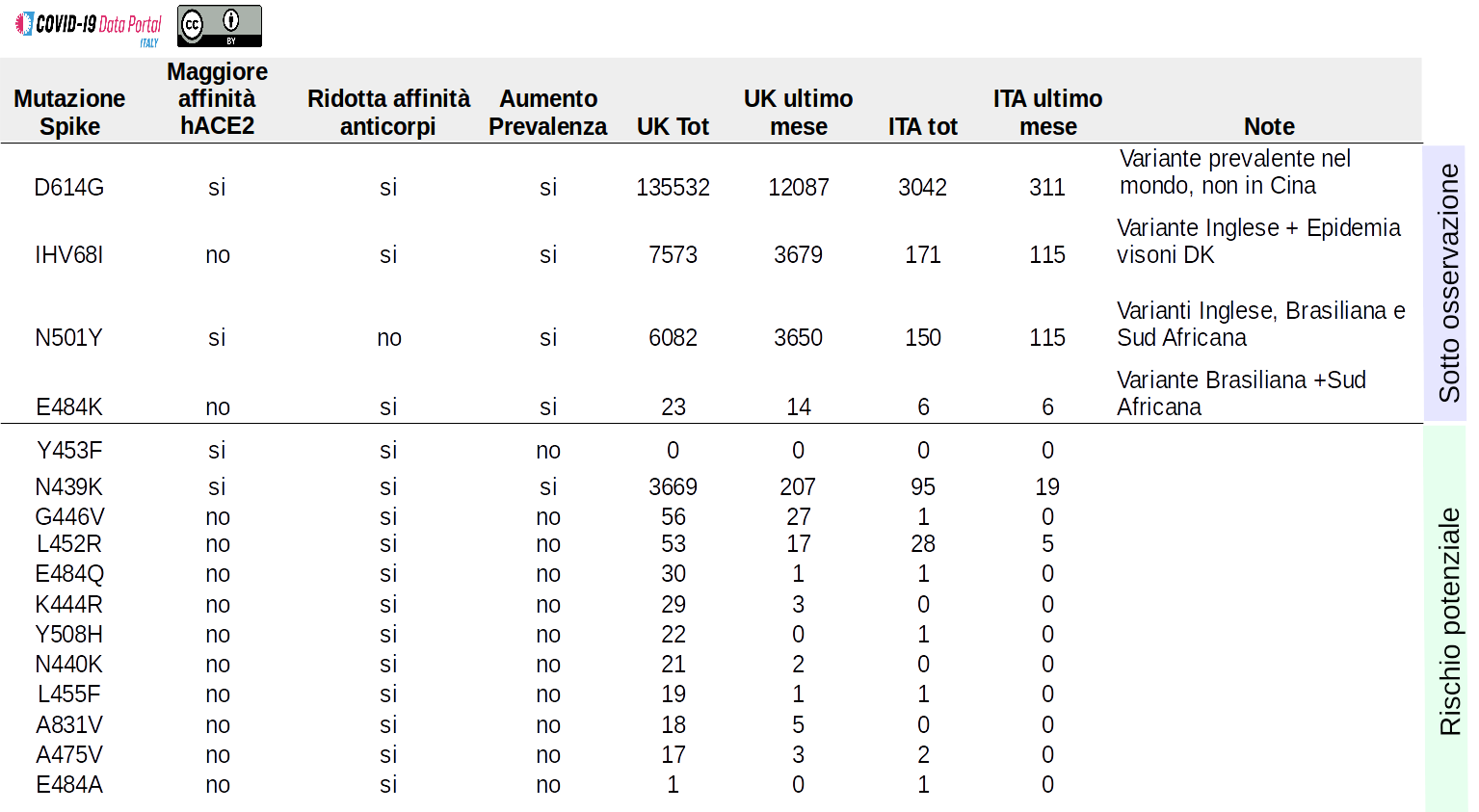
La mutazione D614G non era presente quando il SARS-CoV-2 è stato identificato per la prima volta, ma ora è quasi onnipresente. Si trova in numerose varianti e sembra essere associata ad un moderato aumento della trasmissibilità di SARS-CoV-2 [4].
La mutazione N501Y inizialmente identificata nel Regno Unito [5], è associata a diverse varianti del virus ritenute più contagiose, come la variante inglese (B.1.1.7), quella Brasiliana (P.1) e quella Sudafricana (501Y.V2) [6]. Si ritiene che questa mutazione possa favorire il legame del virus con la proteina recettore umana, aumentandone la trasmissibilità.
E484K è una mutazione specifica della variante Brasiliana (P.1) e di quella Sudafricana (501Y.V2). Studi preliminari suggeriscono come questa mutazione sia in grado di diminuire la capacità di alcuni anticorpi di riconoscere il virus [7]. Analisi preliminari, effettuate in Brasile suggeriscono una maggiore prevalenza di questa mutazione nei casi di re-infezione. Questi dati sono comunque da ritenersi preliminari [8].
IHV68I è una delezione, una mutazione associata con la perdita di 2 aminoacidi, nella glicoproteina Spike, che è comparsa diverse volte in maniera “indipendente” nel genoma di diverse varianti di SARS-CoV-2. E’ stata identificata per la prima volta in pazienti immuno-soppressi trattati con anticorpi monoclonali [9], ma è anche associata ad un evento di “salto di specie” da uomo e visone e poi da visone nuovamente a uomo che ha causato un’epidemia in Danimarca [10]. Questa delezione è stata riscontrata anche nella variante inglese B.1.1.7. Si ritiene diminuire la capacità dei nostri anticorpi di riconoscere il virus [11].
Descrizione delle mutazioni che rappresentano un rischio potenziale
Le mutazioni indicate come “rischio potenziale” non sono associate a nessuna particolare variante di interesse epidemiologico del virus, ma hanno caratteristiche particolari che le rendono degne di nota. Vengono riportate solo per completezza di informazione e per favorire futuri confronti. Al momento attuale non costituiscono fonte di preoccupazione e/o allarme.
Tra queste, l’unica al momento degna di nota, per ora è la mutazione N439K. Studi preliminari indicano che questa particolare mutazione è in grado sia di migliorare la capacità di SARS-CoV-2 di riconoscere la proteina recettore hACE2, che al contempo di rendere il virus meno riconoscibile dal sistema immunitario umano [12]. Di recente si sono riscontrate diverse varianti di SARS-CoV-2 che portano nel loro genoma sia N439K che IHV68I [9] . Le implicazioni funzionali non sono al momento chiare.
Fonte: https://www.gisaid.org/ (dati genomici)
Riferimenti bibliografici
[1] “Weekly epidemiological update -27January 2021” (https://www.who.int/publications/m/item/weekly-epidemiological-update—27-january-2021)
[2] “European Centre for Disease Prevention and Control. Risk related to spread of new SARS-CoV-2 variants of concern in the EU/EEA, first update –21 January 2021. ECDC: Stockholm; 2021.” (https://www.ecdc.europa.eu/sites/default/files/documents/COVID-19-risk-related-to-spread-of-new-SARS-CoV-2-variants-EU-EEA-first-update.pdf)
[3] “European Centre for Disease Prevention and Control. Sequencing of SARS-CoV-2: first update. 18 January 2021. ECDC: Stockholm; 2021. (https://www.ecdc.europa.eu/sites/default/files/documents/Sequencing-of-SARS-CoV-2-first-update.pdf)
[4] Korber, B., et al., Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell, 2020. 182(4): p. 812-827.e19
[5] Rambaut, A., et al. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. 2020; Available at: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineagein-the-uk-defined-by-a-novel-set-of-spike-mutations/563.
[6] Tegally, H., et al., Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv, 2020: p.2020.12.21.20248640.
[7] Weisblum, Y., et al., Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. bioRxiv, 2020: p. 2020.07.21.214759.
[8] Resende, P.C., et al. Spike E484K mutation in the first SARS-CoV-2 reinfection case confirmed in Brazil, 2020. 2021; Available at: https://virological.org/t/spike-e484k-mutation-in-the-first-sars-cov-2-reinfection-case-confirmed-in-brazil-2020/584
[9] Kemp, S., et al., Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/V70. bioRxiv, 2020: p. 2020.12.14.422555.
[10] Fonager J., et al., Working paper on SARS-CoV-2 spike mutations arising in Danish mink, their spread to humans and neutralization data. Available at https://files.ssi.dk/Mink-cluster-5-short-report_AFO2. 2020.
[11] Haynes, W.A., et al., Impact of B.1.1.7 variant mutations on antibody recognition of linear SARS-CoV2 epitopes. medRxiv, 2021: p. 2021.01.06.20248960.
[12] Thomson, E.C., et al., The circulating SARS-CoV-2 spike variant N439K maintains fitness while evading antibody-mediated immunity. Cell, 2021. Epub ahead of print https://doi.org/10.1016/j.cell.2021.01.037.